Search results
Search for "computational chemistry" in Full Text gives 40 result(s) in Beilstein Journal of Organic Chemistry.
Biphenylene-containing polycyclic conjugated compounds
Beilstein J. Org. Chem. 2023, 19, 1895–1911, doi:10.3762/bjoc.19.141

- phenomenon is a result of non-radiative deactivation was corroborated by detailed computational chemistry studies. In continuation of the aforementioned work by Wagner and co-workers [49], they have synthesized also "v" and "z"-shaped POAs, wherein biphenylene groups are angularly incorporated into the 1,4























Thienothiophene-based organic light-emitting diode: synthesis, photophysical properties and application
Beilstein J. Org. Chem. 2023, 19, 1849–1857, doi:10.3762/bjoc.19.137

- absorption and emission maxima of 411 and 520 nm, respectively, with a mega Stokes shift of 109 nm and fluorescence quantum yields both in the solid state (41%) and in solution (86%). The optical properties were supported by computational chemistry using density functional theory for optimized geometry and
- . The high thermal stability is profitable for the preparation of stable and durable OLED devices. Computational chemistry Ground-state geometry optimization of DMB-TT-TPA (8) was performed using density functional theory (DFT) calculations with the Gaussian 16 software at the B3LYP/6-31G (d,p) level







Quinoxaline derivatives as attractive electron-transporting materials
Beilstein J. Org. Chem. 2023, 19, 1694–1712, doi:10.3762/bjoc.19.124

- compound with the donor polymer PM6 to create BHJ nanoparticles and employed it in the hydrogen evolution reaction. This approach substantially reduced trap density, increasing the hydrogen evolution rate by 2–3 times compared to conventional inorganic/organic hybrid photocatalysts [30]. Computational
- chemistry offers a cost-effective and time-efficient means of screening and selecting promising candidates for experimental exploration. Bhattacharya et al. employed density functional theory (DFT) approach to explore structural modulation for tuning the optoelectronic properties of Qx13. Their designed









Unraveling the role of prenyl side-chain interactions in stabilizing the secondary carbocation in the biosynthesis of variexenol B
Beilstein J. Org. Chem. 2023, 19, 1503–1510, doi:10.3762/bjoc.19.107

- , computational chemistry including DFT [5][6][7][8][9], QM/MM [10][11][12][13][14][15][16] and QM/MM MD [14][15][16][17] calculations have been used for the biosynthetic studies of terpene/terpenoids [18]. Terpene-forming reactions, which involve various types of carbocation species stabilized by





Total synthesis: an enabling science
Beilstein J. Org. Chem. 2023, 19, 474–476, doi:10.3762/bjoc.19.36

- [12], as illustrated in this thematic issue with the synthesis of pheromones [16]. This requires permanent technological progress. Thus, the recent boom of artificial intelligence, machine learning, and computational chemistry for retrosynthetic analyses and beyond foreshadows a renewed interest in


Recommendations for performing measurements of apparent equilibrium constants of enzyme-catalyzed reactions and for reporting the results of these measurements
Beilstein J. Org. Chem. 2023, 19, 303–316, doi:10.3762/bjoc.19.26

- reactions that involve the same or a similar change in chemical bonding, e.g., by using group contribution methods [32][33][34][35]. And, one should not overlook the use of computational chemistry as a means to obtain the desired property values. Finally, there is a close relationship between equilibrium

Experimental and theoretical studies on the synthesis of 1,4,5-trisubstituted pyrrolidine-2,3-diones
Beilstein J. Org. Chem. 2022, 18, 1140–1153, doi:10.3762/bjoc.18.118

- Nguyen Tran Nguyen Vo Viet Dai Nguyen Ngoc Tri Luc Van Meervelt Nguyen Tien Trung Wim Dehaen Department of Chemistry, University of Science and Education, the University of Da Nang, Ton Duc Thang 459, 550000 Da Nang, Viet Nam Laboratory of Computational Chemistry and Modelling, Faculty of Natural












Enzymes in biosynthesis
Beilstein J. Org. Chem. 2022, 18, 1131–1132, doi:10.3762/bjoc.18.116

- novel products [7]. An alternative approach is offered by computational chemistry, which is ideally performed in combination with experimental verification of the computational results, e.g., through the enzymatic conversion of isotopically labelled compounds [8]. This thematic issue will cover all

The enzyme mechanism of patchoulol synthase
Beilstein J. Org. Chem. 2022, 18, 13–24, doi:10.3762/bjoc.18.2

- results from these experiments are contradictory. The present work reports on a reinvestigation of patchoulol biosynthesis by isotopic labelling experiments and computational chemistry. The results are in favour of a pathway through the neutral intermediates germacrene A and α-bulnesene that are both











Direct synthesis of anomeric tetrazolyl iminosugars from sugar-derived lactams
Beilstein J. Org. Chem. 2021, 17, 115–123, doi:10.3762/bjoc.17.12

- an attempt at resolving this problem by means of the electronic circular dichroism (ECD) technique. We recorded an ECD spectrum of both compounds and compared it with simulated spectra, generated for both possible diastereomers (2-(R) and 2-(S)) using computational chemistry software. Unfortunately













Synthesis of purines and adenines containing the hexafluoroisopropyl group
Beilstein J. Org. Chem. 2020, 16, 2739–2748, doi:10.3762/bjoc.16.224

- supplementary publication nos. CCDC #1998565−1998569. Computational chemistry Computational optimization of 8a and 8b structures was performed with Gaussian 9 software. Density functional theory at the B3LYP level, with a 6-311g++ basis set (Co phase) was employed. Reaction of purine (2) and 1 A mixture of 1.2











Comparative ligand structural analytics illustrated on variably glycosylated MUC1 antigen–antibody binding
Beilstein J. Org. Chem. 2020, 16, 2540–2550, doi:10.3762/bjoc.16.206

- ] and MDTraj [39]) which are available as computational chemistry analysis tools in Galaxy [6], and these were used to analyze the molecular dynamics trajectories. The root-mean-square deviation (RMSD) is calculated to measure the stability and conformation of a set of selected atoms. The RMSD is a
- . Acknowledgements We would like to acknowledge the Galaxy community, the Galaxy Europe team, and the Galaxy computational chemistry team on GitHub. We thank the University of Cape Town eResearch (for support and use of the ilifu data centre) and the Centre for High Performance Computing (for the use of their GPU






Synthesis, docking study and biological evaluation of ᴅ-fructofuranosyl and ᴅ-tagatofuranosyl sulfones as potential inhibitors of the mycobacterial galactan synthesis targeting the galactofuranosyltransferase GlfT2
Beilstein J. Org. Chem. 2020, 16, 1853–1862, doi:10.3762/bjoc.16.152

- mechanism studies using computational chemistry methods. The probable reaction mechanisms were studied by hybrid DFT QM/MM molecular dynamics simulations [11] where the possible transition state (TS) structures were localized. The observation of the possible TS structure opens the opportunities for the in
- synthesized. We performed docking studies of these compounds into the active site of GlfT2 by computational chemistry methods. Although the docking study showed good binding affinities of the prepared compounds towards the GlfT2 active site, their biological evaluation revealed a very poor effect on the










One-pot synthesis of isosorbide from cellulose or lignocellulosic biomass: a challenge?
Beilstein J. Org. Chem. 2020, 16, 1713–1721, doi:10.3762/bjoc.16.143

- particular, in situ kinetic studies by spectroscopic methods [31] correlated to computational chemistry approaches [32] might reveal specific interaction between reactants and catalysts as well as solvent effects at work in such a complex system that should provide an understanding of the underlying reason








In silico rationalisation of selectivity and reactivity in Pd-catalysed C–H activation reactions
Beilstein J. Org. Chem. 2020, 16, 1465–1475, doi:10.3762/bjoc.16.122
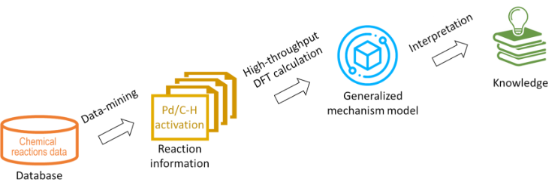
- transition metal coordination sphere; the energy of a new M–C bond formed and the thermodynamic stability of organometallic product. With new developments in computational chemistry, mechanistic studies using density functional theory (DFT) provide valuable insights into the reactivity of organometallic



Photophysics and photochemistry of NIR absorbers derived from cyanines: key to new technologies based on chemistry 4.0
Beilstein J. Org. Chem. 2020, 16, 415–444, doi:10.3762/bjoc.16.40
























Synthesis and characterization of bis(4-amino-2-bromo-6-methoxy)azobenzene derivatives
Beilstein J. Org. Chem. 2019, 15, 3000–3008, doi:10.3762/bjoc.15.296

- absorption wavelengths of possible derivatives. Based on these considerations, we carried out the synthesis and photochemical characterization of compounds 4 and 5. Results and Discussion Computational chemistry Calculations were performed using density functional theory (DFT) methods (B3LYP/6-31+G**) to






Pigmentosins from Gibellula sp. as antibiofilm agents and a new glycosylated asperfuran from Cordyceps javanica
Beilstein J. Org. Chem. 2019, 15, 2968–2981, doi:10.3762/bjoc.15.293

- , Narathiwat 96000, Thailand Computational Chemistry Laboratory, Chemistry Department, Faculty of Science, Minia University, 61519, Egypt National Centre for Genetic Engineering and Biotechnology (BIOTEC), NSTDA, 113 Thailand Science Park, Phahonyothin Road, Klong Nueng, Klong Luang, Pathum Thani 12120







Dyes in modern organic chemistry
Beilstein J. Org. Chem. 2019, 15, 2798–2800, doi:10.3762/bjoc.15.272

- theoretical and computational chemistry [20]. In the same way, essential dye properties, such as aggregation [21][22] or solvatochromism [23], are still in the focus of current physical organic research. The analytical sciences also depend very much on the availability of appropriate dyes with characteristic
The phenyl vinyl ether–methanol complex: a model system for quantum chemistry benchmarking
Beilstein J. Org. Chem. 2018, 14, 1642–1654, doi:10.3762/bjoc.14.140

- progress made in experiments and theory/computational chemistry, there is still a need for improvement and benchmarking [7]. Many aromatic solute–solvent complexes have been studied in the gas phase (cf. [8][9][10] and references therein). Studied systems involving methanol as attached solvent molecule







A permutation approach to the assignment of the configuration to diastereomeric tetrads by comparison of experimental and ab initio calculated differences in NMR data
Beilstein J. Org. Chem. 2017, 13, 2478–2485, doi:10.3762/bjoc.13.245

- capable software has led to inclusion of computational chemistry into the organic laboratory practice [2]. This provided a powerful tool to augment the interpretation of experimental data including NMR results [3][4][5][6][7][8][9][10]. Comparisons of experimental and theoretical spectra for the pairs of



Theoretical simulation of the infrared signature of mechanically stressed polymer solids
Beilstein J. Org. Chem. 2017, 13, 1710–1716, doi:10.3762/bjoc.13.165

- is influenced as well [30], resulting in the observed force-dependent shift of the infrared bands and changes in the intensity. Computational chemistry has proven to be an indispensable tool in the analysis of mechanochemical phenomena of organic molecules, polymers and mechanophores [5][6][31][32









Characterization of the synthetic cannabinoid MDMB-CHMCZCA
Beilstein J. Org. Chem. 2016, 12, 2808–2815, doi:10.3762/bjoc.12.279

- chiral HPLC. Supporting Information Supporting Information File 383: NMR spectra, UV and ECD spectra, IR and VCD spectra, HPLC/ESI-MSn, chiral HPLC, and computational chemistry. Supporting Information File 384: Chemical information file of compound (S)-3. CCDC 1521512 contains the supplementary








Thiophene-forming one-pot synthesis of three thienyl-bridged oligophenothiazines and their electronic properties
Beilstein J. Org. Chem. 2016, 12, 2055–2064, doi:10.3762/bjoc.12.194

- proceeds with lower oxidation potentials and consistently reversible oxidations can be identified. The Stokes shifts are large and substantial fluorescence quantum yields can be measured. Computational chemistry indicates lowest energy conformers with sigmoidal and helical structure, similar to
- and typical for many 3-(hetero)arylphenothiazines the Stokes shifts are large and substantial fluorescence quantum yields can be measured. Computational chemistry supports lowest-energy conformers with sigmoidal and helical structure, similar to oligophenothiazines. Furthermore, TD-DFT and even








Correction: Effective in silico prediction of new oxazolidinone antibiotics: force field simulations of the antibiotic–ribosome complex supervised by experiment and electronic structure methods
Beilstein J. Org. Chem. 2016, 12, 608–610, doi:10.3762/bjoc.12.59
- ; computational chemistry; drug design; molecular recognition; relaxed force constants; Our original publication contains an erratic number of predicted antibiotic structures in Scheme 2. With this Erratum we provide the corrected Scheme 2. Scheme 2 in the original article: Predicted new linezolid-like
